The U.S. Food and Drug Administration (FDA) is making efforts to modernize both the 510(k) approval process for medical devices and study designs for drugs and biologics.
On September 7, 2023, the FDA Center for Devices and Radiological Health (CDRH) issued three draft device guidances:
1) Best Practices for Selecting a Predicate Device to Support a Premarket Notification [510(k)] Submission;
2) Recommendations for the Use of Clinical Data in Premarket Notification [510(k)] Submissions; and
3) Evidentiary Expectations for 510(k) Implant Devices.
On September 8, 2023, the FDA issued an updated guidance on device biocompatibility, Use of International Standard ISO 10993-1, “Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process,” with a special focus on biocompatibility of certain devices in contact with intact skin.
Additionally, on August 30, 2023, the FDA Center for Drug Evaluation and Research (CDER) issued final guidance for drug and biologic study designs titled, Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drugs and Biological Products.
And on September 19, 2023, CDER and the Center for Biologics Evaluation and Research (CBER) released the draft guidance, Demonstrating Substantial Evidence of Effectiveness With One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence.
Each guidance is described below.
Device Guidances
Best Practices for Selecting a Predicate Device to Support a Premarket Notification 510(k) Submission
As background, predicate devices are marketed Class II devices that serve as benchmarks for evaluating the safety and effectiveness of new medical devices seeking FDA clearance for U.S. commercialization. Manufacturers submit a 510(k) application that compares their new device to a predicate in terms of intended use and technological characteristics. A successful demonstration of substantial equivalence to a predicate device results in FDA clearance for market entry.
The draft guidance proposes best practice considerations for selecting a predicate device to support a 510(k) submission. Specifically, the guidance states a sponsor should select a predicate device that: 1) was cleared using well-established methods; 2) meets or exceeds expected safety and performance (which can be confirmed by searching for any reported injury, deaths, or malfunctions using various FDA databases, such as the Medical Device Reporting Database); 3) is without unmitigated use-related or design-related safety issues (which can be confirmed by searching the Medical Device Safety and CBER Safety & Availability (Biologics) websites to assess whether any of the valid predicate devices have an associated use-related or design-related safety issue); and 4) is without an associated design-related recall (which can be confirmed by searching the Medical Device Recalls Database to assess whether any of the valid predicate devices have an associated recall).
The draft guidance also advises submitters to include in their 510(k) submission an explanation of how they applied the best practices outlined in the guidance when selecting the predicate device to support their submission. If a suitable predicate device meeting these best practices isn't available, the FDA recommends that submitters describe in their 510(k) submission how any known concerns related to the valid predicate device have been addressed with the subject device, such as through design modifications or performance testing. Additionally, the FDA suggests summarizing in the 510(k) Summary section of the marketing application how the best practices were employed in choosing the predicate device.
Recommendations for the Use of Clinical Data in Premarket Notification 510(k) Submissions
The FDA stated this draft guidance is meant to outline its current perspective on the use of clinical data in 510(k) submissions and aims to improve the predictability, consistency, and transparency of the 510(k) Program. The guidance provides clarification and additional context for situations where clinical data might be necessary to demonstrate substantial equivalence, as initially outlined in the authoritative guidance, "The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]" (the "510(k) Program Guidance"). In the 510(k) Program Guidance, the FDA detailed common scenarios where clinical data may be required in a 510(k) submission; this new guidance further elaborates on these scenarios and introduces an additional scenario—when a newly identified or increased risk for the predicate device suggests clinical data may be needed for the new device in order to determine substantial equivalence. The guidance also provides additional examples meant to illustrate when clinical data may or may not be required.
The guidance identifies four broad considerations to be used by industry and the FDA to help determine whether clinical data may be necessary to demonstrate that a new device is substantially equivalent to a predicate device: 1) there are differences in the indications for use of the new device compared to the predicate device, and clinical data may be necessary to determine substantial equivalence (e.g., differences in the patient population, disease, or pathology); 2) there are differences in the technological characteristics of the new device compared to the predicate device, and clinical data may be required to determine substantial equivalence (e.g., significant change in materials, device design, or energy source); 3) in cases where substantial equivalence cannot be determined through nonclinical testing such as analytical, bench, and/or animal testing (e.g., there is no model available, the available models are not adequate because the model has certain limitations that do not allow for an adequate assessment, or the model may not be predictive of clinical outcomes); and 4) if a newly identified or increased risk associated with the predicate device suggests that clinical data may be required for the new device to establish substantial equivalence (e.g., unexpected adverse events are reported once the device is more widely distributed and used in clinical practice).
Evidentiary Expectations for 510(k) Implant Devices
Per the FDA, the purpose of this guidance is to assist the industry in designing and conducting appropriate performance testing, which may be necessary to support 510(k) submissions for implant devices. First, the guidance recommends that submitters evaluate general considerations including the indications for use, the intended duration of the implant, and the anticipated patient and physician experience with the implant. The guidance also recommends that submitters consider nine nonclinical issues: 1) biocompatibility; 2) sterility and shelf life; 3) reprocessing and cleaning; 4) software and cybersecurity; 5) electrical safety and electromagnetic compatibility; 6) magnetic resonance compatibility; 7) other nonclinical performance testing; 8) animal testing); and 9) device design considerations.
The guidance further recommends that submitters take human factors into consideration particularly with usability. As part of their design controls, manufacturers are advised to conduct a thorough examination of risks associated with device usage. This examination should consider the specific risks linked to device use and the steps taken to minimize these risks. Since accurately gauging the likelihood of use errors can be challenging, and because many such errors may only become apparent during simulated or real device use, the degree of potential harm becomes a more meaningful factor in deciding whether actions should be taken to eliminate or reduce resulting harm. If the analysis of use-related risks suggests that use errors could potentially result in serious harm to the patient or device user, the FDA strongly recommends the implementation of appropriate human factors/usability (HF/U) engineering processes in accordance with the FDA guidance, “Applying Human Factors and Usability Engineering to Medical Devices.”
The guidance also makes reference to the above guidance titled, “Recommendations for the Use of Clinical Data in Premarket Notification 510(k) Submissions,” in addressing some scenarios where clinical data may be needed to support a substantial equivalence determination for 510(k) implant devices. This includes the collection, analysis, and integration of patient experience data for implants; patient experience data includes patient preference information (PPI) and patient-reported outcomes. PPI may be used to help understand the relative value or the tradeoffs patients are willing to make among different benefits and risks associated with their condition and its diagnosis or management.
Finally, the guidance states that, to the extent not already required under 21 C.F.R. Part 801 regarding device labeling or by applicable special controls, the FDA recommends that all implants be accompanied by labeling that includes information on device operation, implantation instructions, and implant removal if the device is intended to be removed. Additionally, the FDA recommends that manufacturers provide detailed implant identification cards for use by patients or their caregivers, including information such as model name, implant location, how to report malfunctions or other adverse events, and details about composition and patient contacting materials.
The guidance notes that some of these recommendations, such as those related to identifying and mitigating specific risks associated with implants, may have relevance beyond the context of preparing a 510(k) submission, and that these recommendations can be beneficial throughout the entire product lifecycle.
On October 26, 2023, the FDA will host a webinar for industry and other stakeholders interested in learning more about these draft guidances and the FDA's ongoing efforts to modernize the 510(k) Program. Comments for the three 510(k) guidances are due on December 6, 2023.
Use of International Standard ISO 10993-1, “Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process”
This guidance has traditionally been used to prepare Premarket Applications, Humanitarian Device Exceptions, Investigational Device Applications, Premarket Notifications (510(k)s), and De Novo requests for medical devices that come into direct contact or indirect contact with the human body in order to determine the potential for an unacceptable adverse biological response resulting from contact of the component materials of the device with the body.
This most significant development in this recent guidance update is contained in Attachment G of the guidance, which describes a new policy for certain devices that contact intact skin. The policy applies to devices that are 1) medical devices or components that contact intact skin surfaces only; 2) for limited (<=24 hour), prolonged (>24 hours to 30 days), and long term (>30 days) durations of contact, including repeat use devices; and 3) are composed of materials made from certain synthetic polymers or certain natural fabrics, as listed in Attachment G.
Medical devices, components, or materials excluded from this policy are described in Table G.1 below:
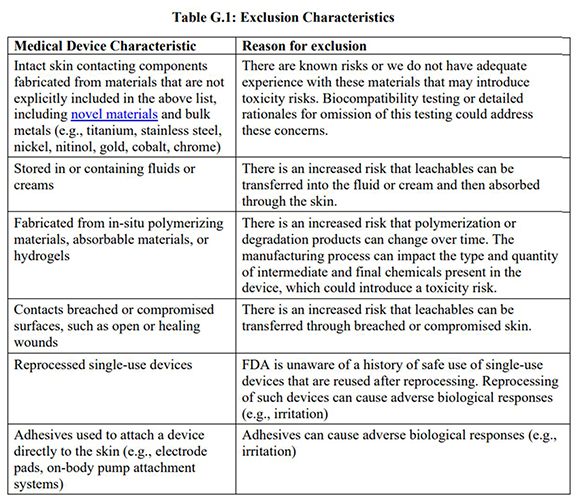
The policy for biocompatibility of certain devices in contact with intact skin described in Attachment G describes a “least burdensome” approach that recommends specific material information to be included in a premarket submission in lieu of biocompatibility testing. This approach also supports the principles of the “3Rs,” to replace, reduce, and/or refine animal use in testing when feasible. Attachment G also includes recommendations for what information and labeling should be provided in premarket submissions for device types within the scope of the policy outlined in Attachment G. Among other things, that information consists of 1) a list of all materials (including color additives) used to fabricate the device or component with only direct or indirect skin contact; 2) a statement confirming that the device materials are listed in Attachment G for inclusion in the policy, and have a documented history of safe use in legally marketed medical devices in contact with intact skin (e.g., through Medical Device Reporting (MDR) analysis, literature search); and 3) a statement confirming that none of the exclusions listed in Table G.1 above apply.
On October 12, 2023, the FDA will host a webinar for industry and other interested stakeholders to discuss the final guidance, with particular focus on the policy in Attachment G.
Drug and Biologic Final Guidances
Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drugs and Biological Products
This final guidance from CDER and CBER focuses on providing guidance on Real-World Data (RWD) use in noninterventional (observational) designs. The FDA defines RWD as data relating to patient health status and/or the delivery of healthcare routinely collected from a variety of sources; and Real-World Evidence (RWE) as the clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD. Various sources of RWD can be analyzed in noninterventional studies, including registries, electronic health records (EHRs), and medical claims.
As background, the 21st Century Cures Act (Cures Act), which was signed into law on December 13, 2016, in part added section 505F to the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 355g). Pursuant to this section, the FDA created the RWE Program as a framework to evaluate the potential use of RWE in regulatory decision-making. This guidance is part of a series of guidances that the FDA has already published, or plans to publish, as part of the agency’s RWE Program and in support of the Cures Act.
The guidance outlines the FDA’s general expectations for study conduct for sponsors submitting new drug applications or biologics license applications using RWD in noninterventional (observational) studies. Noninterventional studies analyze data reflecting the use of a marketed drug administered in routine medical practice, according to a medical provider’s clinical judgment and based on patient characteristics, rather than assignment of a participant to a study arm according to a research protocol. Accordingly, the FDA acknowledges in the guidance that noninterventional studies are not clinical investigations as defined under § 312.3 and do not require an IND.
The guidance divides its considerations for non-interventional studies involving the use of RWD into five categories: 1) transparency regarding data collection and analysis; 2) access to RWD; 3) study monitoring; 4) safety reporting; and 5) other sponsor responsibilities. Examples from each category are listed below:
- In the transparency category, the guidance says that sponsors planning to use a noninterventional study to support a marketing application should engage with the FDA early in the drug development process using an appropriate regulatory pathway (e.g., requesting a Type C meeting through an existing IND for the product). Additionally, the guidance states that sponsors should enable and maintain audit trails of data, starting from extracting RWD sources through maintenance and retention of dataset(s), and that this process should include the tracking of user access, data changes, changes to the protocol, and analyses performed.
- In the access category, the guidance notes that, if certain RWD are owned and controlled by other entities, sponsors should have agreements in place with those entities to ensure that relevant patient-level data can be provided to the FDA and that source data necessary to verify the RWD are made available for inspection as applicable.
- In the study monitoring category, the guidance states that sponsors should, at a minimum ensure that: the RWD required by the protocol are accurate and consistent with the source records; the prespecified plans, protocol, and study procedures were followed; and deviations from the plans, protocol, and procedures are identified, documented, evaluated, and remediated.
- In the safety reporting category, the guidance says that, if a sponsor identifies adverse events that are subject to post-marketing reporting requirements during the course of conducting a noninterventional study, such events must be reported in accordance with applicable post-marketing reporting requirements.
- Finally, in the catchall category, the guidance says that the FDA expects that the sponsor will retain and make available to the FDA upon request a log of any researcher or researchers who have significant involvement in the design or conduct of the study, and that the log should contain information on researchers, including their name, affiliations, description of roles or activities performed, and qualifications regarding education, training, and experience to perform the proposed study role.
Demonstrating Substantial Evidence of Effectiveness Based on One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence
This draft guidance complements the 2019 draft guidance, “Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products” (the 2019 Effectiveness draft guidance), and the 1998 guidance, “Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products” (the 1998 Effectiveness guidance). Although the FDA’s evidentiary standard has not changed since 1998, the FDA said there is a need for additional guidance to describe how one adequate and well-controlled clinical investigation and confirmatory evidence can be used to meet the substantial evidence requirement for demonstrating human drug and biological product effectiveness—and this new draft guidance serves that purpose.
The substantial evidence of effectiveness standard, as defined in the Food Drug and Cosmetic Act (FD&C Act), includes both the quantity and quality of evidence. In the 2019 Effectiveness draft guidance, certain aspects of adequate and well-controlled clinical investigations were discussed, specifically focusing on trial design, endpoints, and statistical considerations. The 2023 draft guidance adds that the quality and quantity of confirmatory evidence are also important considerations and should be generated from quality data derived from an appropriate source.
The guidance describes and provides considerations for seven types of confirmatory evidence:
- clinical evidence from a related indication;
- mechanistic or pharmacodynamic evidence;
- evidence from a relevant animal model;
- evidence from other members of the same pharmacological class;
- natural history evidence;
- RWD and evidence; and
- evidence from expanded access use of an investigational drug.
The draft guidance also provides recommended process considerations for sponsors’ discussions with their review divisions. The FDA recommends that sponsors do the following:
- Offer a strong scientific rationale for use of a single clinical study and confirmatory evidence for their specific drug development program;
- describe the expected design of one adequate and well-controlled clinical investigation that the confirmatory evidence will support; and
- discuss the confirmatory evidence they intend to use, including the type (i.e., data source) and quantity of confirmatory evidence that will be included in the application.
The policy in the guidance underscores the FDA’s discretion in whether data from one adequate and well-controlled clinical investigation and confirmatory evidence are sufficient to demonstrate effectiveness. The guidance states that the quantity (e.g., number of sources) of confirmatory evidence necessary to support effectiveness may vary across development programs and the quantity of confirmatory evidence needed in a development program will be impacted by the features of, and results from, the single adequate and well-controlled clinical investigation that the confirmatory evidence is intended to substantiate. Further, it may be possible for a highly persuasive adequate and well-controlled clinical investigation to be supported by a lesser quantity of confirmatory evidence, whereas a less-persuasive adequate and well-controlled clinical investigation may require a greater quantity of compelling confirmatory evidence to allow for a conclusion of substantial evidence of effectiveness. The guidance states that a clinical investigation’s particular set of features (e.g., trial design, endpoints, and statistical considerations) will result in a greater or lesser degree of certainty that the FDA is willing to accept about effectiveness.
Comments on the draft guidance are due on December 18, 2023.
